
In Silico Identification of Potential PD-L1 and VISTA Inhibitors in Ovarian Cancer: A Computational Approach Combining Virtual Screening and Molecular Dynamics Simulations
Authors Info & Affiliations
Abstract
Introduction
Immune checkpoint blockade targeting PD-1/PD-L1 has revolutionized cancer treatment; however, resistance remains a major clinical challenge. V-domain Immunoglobulin Suppressor of T cell Activation (VISTA), a B7 family member with high expression in tumor-infiltrating lymphocytes of ovarian cancer, has emerged as a promising alternative target for immunotherapeutic intervention.
Materials and Methods
We performed in silico screening of 9,397 DrugBank compounds against PD-L1 and VISTA using AutoDock Vina. The top candidates based on docking scores were assessed through 100 ns molecular dynamics simulations, and binding free energies were calculated via MM-PBSA.
Results
DB15637, DB12867, and DB06744 showed the strongest PD-L1 binding affinities (−7.33 to −7.87 kcal/mol) with average RMSD values of 8.89 Å, 8.94 Å, and 7.57 Å, respectively. DB00321 exhibited the highest affinity for VISTA (−7.31 kcal/mol) with an RMSD of 6.18 Å, maintaining stable interactions with key residues throughout the simulation.
Discussion
The identified compounds demonstrated favorable docking scores, dynamic stability, and binding free energies, suggesting their potential as PD-L1 and VISTA inhibitors. Dual checkpoint targeting could enhance antitumor immune responses in ovarian cancer, where both proteins contribute to immune evasion.
Conclusion
This in silico study identified promising candidates for PD-L1 and VISTA inhibition. These findings provide a computational basis for further experimental validation to confirm their therapeutic potential in the treatment of ovarian cancer.
1. INTRODUCTION
Ovarian Cancer (OC) is the leading cause of mortality among gynecological cancers, accounting for approximately 207,300 deaths worldwide in 2020 [1]. The standard treatment approach remains optimal debulking surgery followed by platinum-based chemotherapy [2]. In cases of bulky stage III-IV tumors where complete resection is not achievable, neo-adjuvant chemotherapy, interval debulking surgery, and adjuvant chemotherapy provide an effective alternative with reduced morbidity [3, 4]. Despite an 80% response rate to first-line chemotherapy, the majority of patients experience relapse, leading to a five-year survival rate of only 45% [5]. To improve long-term disease remission, innovative treatment strategies are currently being explored.
Immunotherapy has shown significant promise in the treatment of various cancers, particularly through Immune Checkpoint Inhibitors (ICIs) targeting Cytotoxic T Lymphocyte-Associated protein 4 (CTLA-4) or the PD-L1/PD-1 pathway [6, 7]. PD-L1/PD-1 inhibitors are a class of immunotherapeutic agents designed to block inhibitory signaling pathways mediated by PD-L1/PD-1 interactions, thereby unleashing the antitumor immune response and enhancing the elimination of cancer cells by the immune system. Monoclonal antibodies targeting PD-1, such as pembrolizumab and nivolumab, have demonstrated unprecedented efficacy and durable responses in melanoma, Non-Small Cell Lung Cancer (NSCLC), Renal Cell Carcinoma (RCC), and other solid tumors [8-12]. Similarly, PD-L1 inhibitors, such as atezolizumab and durvalumab, have shown significant clinical benefits in NSCLC, urothelial carcinoma, and triple-negative breast cancer [7, 13]. However, despite their success in several tumor types, the efficacy of anti-PD-1 and anti-PD-L1 in ovarian cancer remains modest. A recent network meta-analysis confirmed the limited benefit of PD-1/PD-L1 inhibitors as monotherapy in ovarian cancer, underscoring the urgent need to explore combination strategies [14].
Among these emerging V-domain Ig Suppressor of T cell Activation (VISTA), encoded by the C10orf54 gene and also referred to as the PD-1 homolog, is a recently identified inhibitory immune checkpoint molecule. It is part of the B7 family and exhibits sequence similarity to both PD-1 and PD-L1 [15, 16]. This protein can inhibit T cell activation when present as a ligand on antigen-presenting cells or as a receptor on T cells [17]. Liu et al. identified a unique role for VISTA in T cell activation, distinct from the PD-1/PD-L1 pathway [18]. This discovery supports the rationale for targeting both the VISTA and PD-1/PD-L1 pathways in cancer treatment.
VISTA inhibitors aim to block the immunosuppressive effects mediated by VISTA; its interaction with T cells reduces immune activation, allowing tumor cells to evade immune detection. By inhibiting VISTA, these agents restore T cell proliferation, cytokine secretion, and cytotoxic activity within the tumor microenvironment. Several approaches are currently under investigation, including monoclonal antibodies, such as CI-8993 and JNJ-61610588, as well as small-molecule compounds, like CA-170, that target both VISTA and PD-L1 [19, 20]. Preclinical murine studies have revealed that dual blockade of VISTA and PD-L1 leads to a synergistic therapeutic effect in models of colon cancer [18]. These studies indicate that VISTA may regulate a distinct immune evasion mechanism, making it a promising target for cancer immunotherapy. Across various malignancies, including Non-Small Cell Lung Cancer (NSCLC), colorectal carcinoma, hepatocellular carcinoma, gastric carcinoma, oral squamous cell carcinoma, acute myeloid leukemia, and gestational trophoblastic neoplasia [21, 22]. Given that VISTA and PD-L1 are highly expressed in ovarian cancer, concurrently targeting both checkpoints may enhance therapeutic efficacy [22, 23].
As an alternative to monoclonal antibodies, small-molecule inhibitors offer several advantages, including better oral bioavailability, improved tumor penetration, fewer side effects, and ease of self-administration. These inhibitors also tend to have a shorter biological half-life and are more cost-effective than mAbs [24, 25]. CA-170 is an orally administered therapeutic agent that directly targets PD-L1, PD-L2, and VISTA. In its Phase I clinical trial, this agent was tested in patients with solid tumors, lymphoma, and other types of cancer [26].
Despite these promising developments, most low molecular weight inhibitors targeting the VISTA and PD-1/PD-L1 pathways are still in the early stages of development, with a strong emphasis on preclinical research. In this study, we conducted a virtual screening of the DrugBank database to identify potential neutralizing agents, applying drug-repositioning principles. Molecular docking enables rapid screening of large compound libraries to predict binding affinities, while molecular dynamics simulations provide insights into the stability and flexibility of protein-ligand complexes in a dynamic environment. Additionally, MM-PBSA calculations offer refined estimates of binding free energies, improving the selection of promising candidates for further validation. These methods are well-established and widely used in drug discovery, and their integrated application here aligns with current state-of-the-art approaches, providing a reliable framework to accelerate the identification of viable therapeutic candidates for ovarian cancer.
2. MATERIALS AND METHODS
2.1. Screening Library Preparation
A total of 9,397 compounds were retrieved from the DrugBank database. The ligands were obtained in 3D SDF format. Polar hydrogen atoms were added to all molecules using OpenBabelGUI tools [27], to ensure an accurate representation of partial charges, which is crucial for understanding ligand-receptor interactions [28].
2.2. Target Proteins Preparation
The crystal structures of the human PD-L1 and human VISTA extracellular domains were retrieved from the Protein Data Bank (PDB) with resolutions of 0.99 Å and 1.85 Å, respectively. The corresponding PDB IDs are 5O45 and 6OIL. AutoDockTools was used to prepare the proteins for molecular docking. Heteroatoms and water molecules were removed, while polar hydrogens and Kollman charges were added to ensure proper docking configurations [29]. The prepared structures were saved in AutoDock PDBQT format. The docking grid box was centered around the protein's known interactive residues: Ile54, Tyr56, Met115, Ala121, Tyr123, Asp122, ILE116, SER117, Tyr for PD-L1 [25, 30], and Arg54, Arg58, Phe62, Gln63 for VISTA [31].
2.3. Virtual Screening
Analyzing the interaction between ligands and proteins provides insights into their potential therapeutic activity. To identify potential inhibitors of PD-L1 and VISTA, molecular docking studies were conducted to screen 9,397 compounds against the targets using AutoDock Vina 1.2.5, which employs a stochastic global optimization algorithm with an exhaustiveness parameter of 8. Each compound was docked independently against both targets with 9 docking runs per ligand to explore multiple binding poses. The best-scoring pose for each ligand was selected based on binding affinity and interaction profile. Docking results were ranked according to the binding energy of the top-scoring conformations.
The best-ranked ligands for each protein were selected for further analysis. Docking results for the selected molecules were visualized with PyMOL, and a ligand interaction diagram was created using LigPlot+.
2.4. Molecular Dynamics Simulation
To evaluate the protein–ligand interaction energy, Molecular Dynamics (MD) simulations were conducted for a duration of 100 ns using GROMACS v.2023.3 [32]. Molecules demonstrating high binding affinity with both proteins were selected for these simulations. Charmm-GUI was employed in the preparation of the MD systems [33]. The CHARMM36m force field was used to describe the interactions between the protein and the solvent, with TIP3P employed as the water model [34]. Ligands were parameterized using the CHARMM General Force Field (CGenFF) via the CHARMM-GUI Solution Builder, ensuring full compatibility with the CHARMM36m force field.
Proteins were solvated within a cubic simulation box with an edge length of 1.0 nm. The system was neutralized to a physiological salt concentration of 0.15 M NaCl by introducing an equal number of positive and negative ions.
Energy minimization was performed using the steepest descent algorithm, followed by equilibration at 300 K for 100 ns in an NVT ensemble using the V-rescale thermostat. Production runs were carried out in an NPT ensemble for 100 ns, with pressure equilibration at 1 atm using the Parrinello–Rahman algorithm [35]. Bond constraints were managed using the LINCS algorithm, with distance cut-offs handled through the Verlet scheme [36].
To better comprehend the structural changes occurring throughout the simulation, various parameters were analyzed, including the Root Mean Square Deviation (RMSD) of atomic coordinates, Root Mean Square Fluctuation (RMSF), and Radius of gyration (Rg). These calculations were performed using the GROMACS software package v.2023.3. The trajectory was processed to eliminate the effects of Periodic Boundary Conditions (PBC).
2.5. MM-PBSA Calculation
MMPBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) is a widely used computational method to estimate the binding free energy of protein-ligand complexes. It combines molecular mechanics energy calculations with solvation energy estimates using the Poisson–Boltzmann (PB) or Generalized Born (GB) models and solvent-accessible surface area (SA) calculations [37]. The gmx_MMPBSA package was employed to compute the binding free energy of protein-ligand complexes [38].
2.6. MM-PBSA Energy Decomposition Calculation
To identify specific amino acid residues contributing to the stabilization of the protein–ligand complexes, a per-residue decomposition of the binding free energy was carried out. This analysis allows the quantification of favorable or unfavorable contributions from individual residues to the overall interaction. The decomposition was performed using the dedicated module in gmx_MMPBSA [39], based on the same simulation trajectories and parameters as the global energy calculations. The results provided a detailed map of key residue-level interactions within the PD-L1 and VISTA binding sites.
3. RESULTS AND DISCUSSION
3.1. Molecular Docking Validation and Virtual Screening
Molecular docking is a computational approach commonly used in structural biology and drug development. It serves as an essential tool for identifying molecules that may bind effectively to target receptors. This technique provides insights into binding mechanisms and interactions between ligands and receptors, thereby facilitating progress in the drug discovery process [40].
After refining the structures of PD-L1 and VISTA, the five highest-ranking ligands from the High-Throughput Virtual Screening (HTVS) were selected based on their optimal docking scores Tables 1 and 2. Compared to CA-170, the drug candidates demonstrated strong binding affinities for PD-L1 and VISTA.
For PD-L1 Table 1, docking scores ranged from −7.87 to −7.00 kcal/mol, while for VISTA Table 2, they ranged from −8.42 to −7.07 kcal/mol, indicating a high binding potential. The top three ligands for each protein were further analyzed, and their interactions were visualized.
|
Drugbank Compound IDs |
Compound | Binding Energy PD-L1, KCAL /MOL |
|---|---|---|
| DB12867 | Benperidol | -7,87 |
| DB06744 | Ginkgolide B | -7,36 |
| DB15637 | Fluzoparib | -7,33 |
| DB11941 | Tasisulam | -7,01 |
| DB11875 | Diindolylmethane | -7,00 |
| CA-170 | - | -5,49. |
|
Drugbank Compound IDs |
Compound | Binding Energy, KCAL /MOL |
|---|---|---|
| DB06744 | Ginkgolide B | -8,42 |
| DB16896 | Racanisodamine | -7,57 |
| DB00321 | Amitriptyline | -7,31 |
| DB11941 | Tasisulam | -7,282 |
| DB07163 | 5-[(2-AMINOETHYL)AMINO]-6-FLUORO-3-(1H-PYRROL-2-YL) BENZO[CD]INDOL-2(1H)-ONE | -7,07 |
| CA-170 | - | -5,95. |
3.2. Receptor–Ligand Interaction Analysis of PD-L1 and VISTA
Our study identified the top five molecules with the lowest docking scores for PD-L1 and VISTA Tables 3 and 4. Interaction analyses of the top three PD-L1-binding compounds Table 3 revealed that DB12867 formed one hydrogen bond with Gln66, DB06744 did not establish any hydrogen bonds, and DB11875 formed one hydrogen bond with Asp122, while CA-170 established four hydrogen bonds with Glu58 and Gln66 Table 3. In addition to hydrogen bonding, all four compounds formed an extensive network of hydrophobic interactions with multiple residues within the PD-L1 binding pocket.
| Drugbank compound IDs | 2-D interaction diagram | Drug groups | Indication |
|---|---|---|---|
| DB12867 |
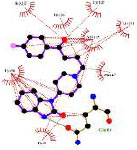
|
Approved | Psychomotor agitation Psychosis Manic Syndromes |
| DB06744 |
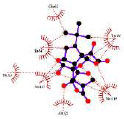
|
Experimental | - |
| DB15637 |
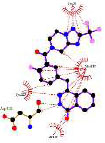
|
Investigational | Metastatic Breast Cancer Metastatic Castration-Resistant Prostate Cancer (mCRPC) Ovarian cancer |
| CA-170 |
|
Investigational | Advanced Solid Tumors or Lymphomas |
| Drugbank compound IDs | 2-D interaction diagram | Drug groups | Indication |
|---|---|---|---|
| DB06744 |
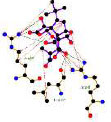
|
Experimental | - |
| DB16896 |
|
Investigational | Jaundice, Obstructive Systemic Inflammatory Response Syndrome (SIRS) |
| DB00321 |
|
Approved | Depression and anxiety Palliative care |
| CA-170 |
|
Investigational | Advanced solid tumors or lymphomas |
In parallel, the interaction pattern analysis of the top three drugs with VISTA (Table 4) suggested that DB06744 formed five hydrogen bonds with Arg55, Arg42, and Leu107. DB16896 formed four hydrogen bonds with Arg42, Arg55, and Arg58, while DB00321 did not form any hydrogen bonds. In contrast, CA-170 formed seven hydrogen bonds with Arg42, Arg55, Leu107, Pro56, and Asn59 Table 4. Similarly, multiple hydrophobic interactions were formed between the binding site residues of the VISTA receptor and each of the selected compounds.
3.3. Molecular Dynamics Simulation Analyses of PD-L1 and VISTA
Molecular Dynamics (MD) simulation is a powerful computational technique in drug design and development. It enables the exploration of molecular interactions at the atomic level, offering deep insights into their structural behavior and dynamic changes over time [41].
By exploring the complexities of MD simulations, our main goal is to uncover the molecular mechanisms underlying these compounds. Examining their atomic-level behavior allows us to better assess their potential as drug candidates. The insights gained from these simulations will not only help optimize these compounds but also support the larger objective of developing safe and effective pharmaceutical treatments. Thus, we aim to investigate the dynamic behavior of the top nine molecules with the lowest docking scores for PD-L1 and VISTA over a 100-ns timescale, analyzing key metrics such as RMSD, RMSF, and Rg.
3.4. RMSD Analysis of Ligand Binding in PD-L1 and VISTA Inhibition
Root Mean Square Deviation (RMSD) is a key metric that quantifies the average atomic displacement over time, comparing the initial reference structure to the subsequent trajectory. It provides insights into the structural stability of both the target receptor and the bound ligand during the simulation.
The RMSD results for the promising ligands of PD-L1 (Fig. 1A) indicate that the reference drug CA-170 has a mean RMSD of 26.69 Å with a Standard Deviation (SD) of 8.21 Å, indicating considerable structural rearrangement during the simulation. The initially high RMSD values observed for the CA-170/PD-L1 complex reflect an early instability of the ligand within the binding site, consistent with the apparent unbinding events during the first 20 ns of the simulation. However, as illustrated in Fig. S1 (Supplementary Material), after this initial phase, the ligand progressively reorients and stabilizes within the PD-L1 binding pocket, maintaining a more consistent conformation for the remainder of the trajectory. In contrast, DB15637 exhibited a favorable RMSD profile, with an average value of 8.89 Å (SD = 1.44 Å). A notable peak around 15 ns, reaching 14 Å, suggests an initial conformational adjustment, followed by sustained stability throughout the simulation, as reflected in the graph. This behavior indicates that DB15637 quickly adapted to its binding site and maintained structural stability thereafter. DB12867 maintained a consistent RMSD profile throughout the simulation with the lowest standard deviation (0.81 Å) and a mean RMSD of 8.94 Å, suggesting limited conformational fluctuation. DB06744 exhibited increased deviations towards the end of the simulation, with an average RMSD of 7.57 Å (SD = 4.49 Å), possibly indicating a rearrangement or partial destabilization at later stages.
For the VISTA-targeting promising ligand (Fig. 1B), the reference drug CA-170 displayed a moderate average RMSD of 15.21 Å (SD = 2.51 Å), while DB00321 displayed the most favorable RMSD profile, with an average RMSD of 6.18 Å (SD of 1.88 Å).
Overall, while RMSD analysis does not directly reflect binding strength, it provides complementary information on the structural dynamics of the complexes. The relatively low and stable RMSD values observed for DB15637, DB12867, and DB00321 suggest that these ligands maintained their positions within the binding site and may warrant further investigation based on their favorable dynamic behavior in conjunction with other energetic and structural analyses.
Ligands that are not shown in the figure exhibited excessive fluctuations during the simulation, indicating unstable binding behavior, and were therefore excluded from further analysis.
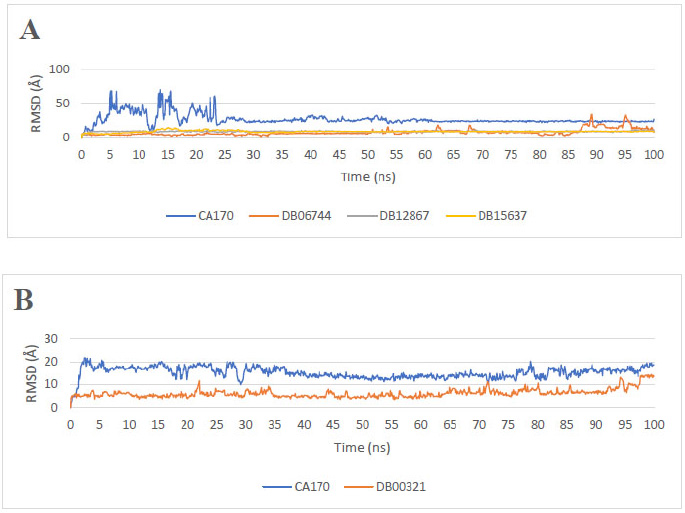
RMSD ligands plot of the repurposed drugs. (A) Reference drug (CA-170) and the three most promising PD-L1 binders. (B) Reference drug (CA-170) and the most promising VISTA binders.
3.5. RMSD Analysis of Protein Conformational Changes in PD-L1 and VISTA
The analysis of the Root Mean Square Deviation (RMSD) profiles presented in Fig. (2A and 2B) provides insights into the conformational stability of PD-L1 and VISTA, both in their unbound states and in the presence of ligands, during molecular dynamics simulations.
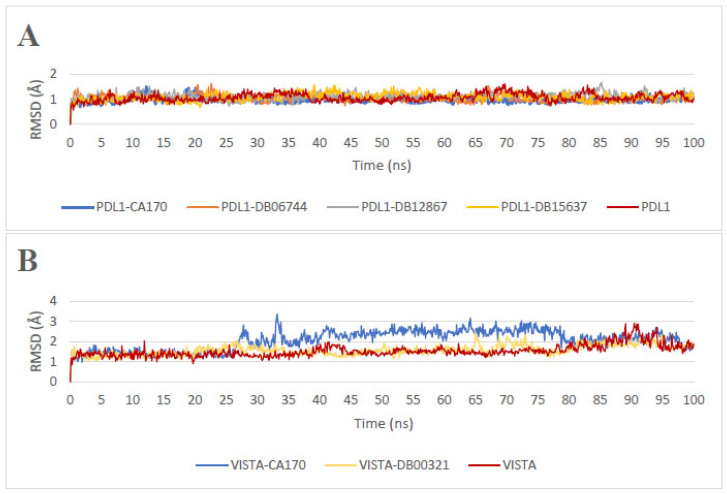
Root Mean Square Deviation (RMSD) profiles: (A) apoprotein PD-L1 and PD-L1 in the presence of ligands, (B) apoprotein VISTA and VISTA in the presence of ligands.
The apoprotein PD-L1 exhibits a mean RMSD of 1.06 Å (SD = 0.16 Å), indicating a relatively stable structure. Upon ligand binding, different trends are observed. In the presence of CA-170, the protein maintains a mean RMSD of 0.98 Å (SD = 0.11 Å), which is lower than that of the apoprotein, indicating a stabilizing effect. Among the other ligands, DB12867 displays the highest RMSD of 1.11 Å (SD = 0.14 Å), suggesting increased protein flexibility, possibly due to conformational rearrangements. DB06744 and DB15637 exhibit RMSD values of 1.03 Å and 1.10 Å, respectively, with SDs of 0.13 Å and 0.16 Å, indicating intermediate stability. These variations suggest that different ligands influence PD-L1 dynamics in distinct ways, potentially affecting their ability to stabilize the protein or modulate its interactions with other molecular partners.
The apoprotein VISTA exhibits a mean RMSD of 1.76 Å (SD = 0.27 Å), indicating a relatively stable structure. However, upon binding to CA-170, the RMSD increases to 2.06 Å (SD = 0.48 Å), suggesting greater structural fluctuations. Notably, a sharp rise in RMSD is observed after approximately 30 ns, exceeding 3.0 Å, which indicates significant conformational changes. In contrast, DB00321 binding results in a more pronounced decrease in RMSD to 1.58 Å (SD = 0.25 Å), accompanied by a lower standard deviation. This suggests that DB00321 stabilizes the VISTA structure, limiting its conformational fluctuations. Such stabilization may be attributed to specific interactions that constrain the dynamic behavior of the protein, thereby reducing its overall flexibility.
3.6. RMSF of PD-L1 and VISTA in Ligand Binding Complexes
The Root Mean Square Fluctuation (RMSF) quantifies the flexibility and movement of atoms in a molecule, offering valuable insights into its structural dynamics [42, 43]. When analyzing the RMSF results for PD-L1 and VISTA, it is crucial to relate these findings to the previous RMSD discussion in order to thoroughly assess the behavior of the compounds during the MD simulation.
For PD-L1 (Fig. 3A), the results indicate that ligand binding significantly affects receptor stability, with notable variations among the complexes. The apoprotein PD-L1 exhibits a mean RMSF of 15.62 Å (SD = 5.29 Å), suggesting greater intrinsic mobility in the absence of a ligand. Ligand binding generally reduces these fluctuations, though differences are observed between complexes. The average RMSF values for PD-L1-CA-170, PD-L1-DB06744, PD-L1-DB12867, and PD-L1-DB15637 are 12.94 Å, 13.89 Å, 13.95 Å, and 15.03 Å, respectively, with corresponding standard deviations of 3.95 Å, 3.82 Å, 4.64 Å, and 5.29 Å. This reduction in flexibility suggests that ligand binding contributes to a more rigid and stable protein conformation, which may influence its functional interactions.
A similar trend is observed for VISTA (Fig. 3B). The apoprotein VISTA exhibits a mean RMSF of 14.09 Å (SD = 4.73 Å), indicating higher mobility. Upon ligand binding, fluctuations are reduced, with mean RMSF values of 12.66 Å (SD = 4.20 Å) for VISTA-CA-170 and 13.49 Å (SD = 4.67 Å) for VISTA-DB00321. This decrease in flexibility suggests that ligand binding stabilizes the protein structure, potentially influencing its interactions. These results collectively highlight the structural dynamics of the binders and their potential applicability in the design of immunotherapeutic drugs.
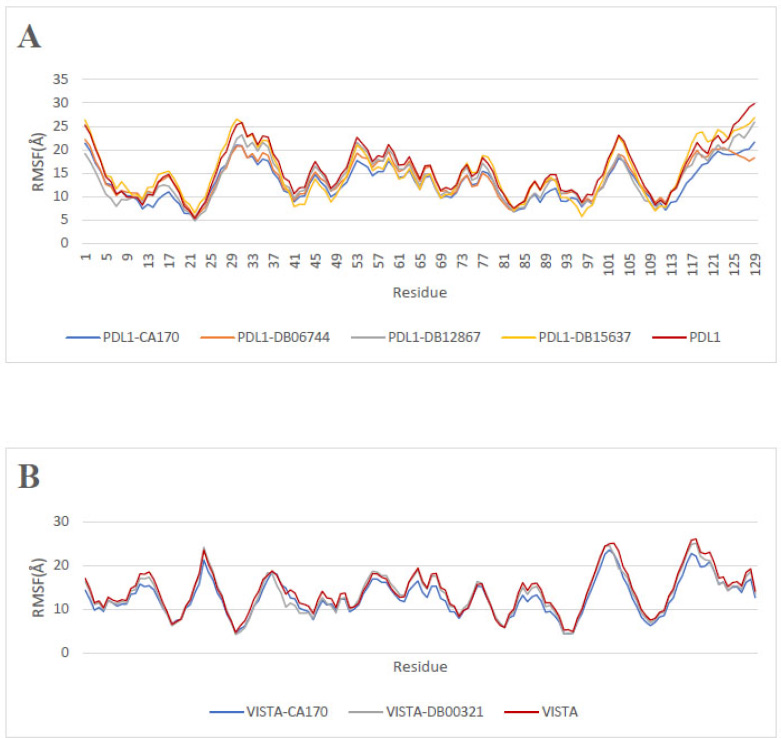
Root Mean Square Fluctuation (RMSF) along the residue positions: (A) apoprotein PD-L1 and PD-L1 in the presence of ligands, (B) apoprotein VISTA and VISTA in the presence of ligands.
3.7. Rg Analysis of PD-L1 and VISTA in Ligand Binding Complexes
The Radius of gyration (Rg) is an indicator of biomolecular structural compactness [44]. The higher the Rg value, the less compact the structure. For interactions with PD-L1 (Fig. 4A), the PD-L1-CA-170 complex exhibits an average Rg value of 16.34 Å (SD = 0.54 Å), which is higher than that of PD-L1 (15.98 Å, SD = 0.42 Å). This increase suggests that CA-170 reduces the compactness of PD-L1, likely due to a decrease in intramolecular interactions. In contrast, the other complexes PD-L1-DB12867, PD-L1-DB15637, and PD-L1-DB06744 display lower Rg values of 15.50 Å, 15.85 Å, and 15.90 Å, respectively, with standard deviations of 0.67 Å, 0.43 Å, and 0.76 Å. These ligands appear to stabilize PD-L1 and promote its compaction, which may indicate an enhanced interaction between the ligand and the protein, leading to reduced structural flexibility.
Regarding the complexes involving the VISTA protein (Fig. 4B), the VISTA-CA-170 complex exhibits an average Rg value of 15.61 Å (SD = 0.15 Å), which is higher than that of unbound VISTA (15.43 Å, SD = 0.10 Å). In contrast, the VISTA-DB00321 complex shows a lower Rg value of 15.34 Å (SD = 0.07 Å), indicating a slightly more compact structure.
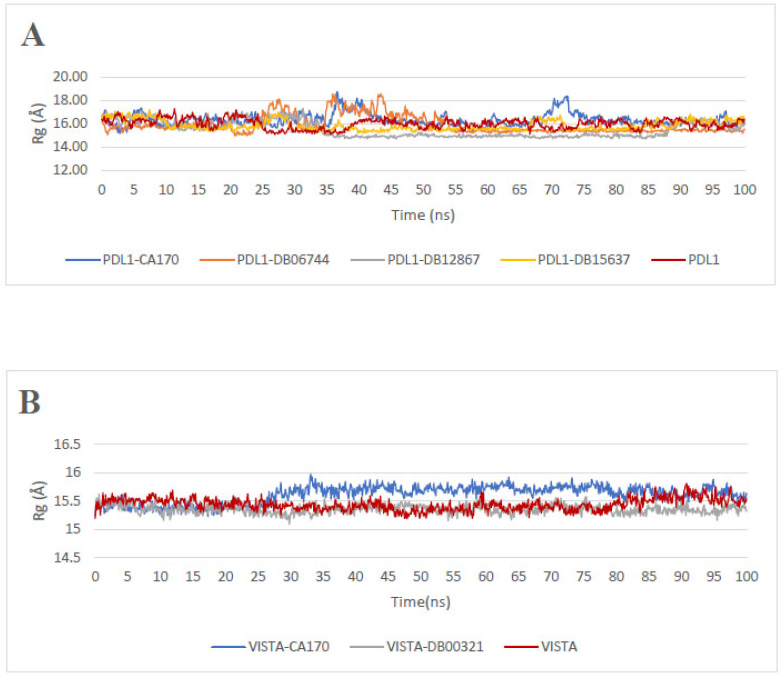
Radius of gyration Rg profiles: (A) apoprotein PD-L1 and PD-L1 in the presence of ligands, (B) apoprotein VISTA and VISTA in the presence of ligands.
3.8. MM-PBSA Analysis of PD-L1 and VISTA in Ligand Binding Complexes
The MD complexes were subjected to MM-PBSA analysis to estimate their binding free energy. MM-PBSA calculations of each complex were performed on the equilibrated last 50 ns of the simulation trajectories in steps of 5 frames. These calculations revealed negative MM-PBSA values for all docked complexes, indicating that ligand binding was favorable due to the negative binding free energy Table 5. The obtained values show significant variations in the affinity of the compounds for PD-L1. DB15637 exhibits the lowest binding energy -19.40 kcal/mol, suggesting a strong affinity for PD-L1, with a SD of 4.44, indicating relatively high interaction stability. Similarly, DB12867 demonstrates good affinity (-12.85 kcal/mol), although with slightly higher variability (SD = 5.07), suggesting moderate interaction stability. DB06744, with a binding energy of -10.78 kcal/mol and a low (SD = 3.82), exhibits relatively stable interactions, although its affinity remains moderate compared to DB15637 and DB12867. CA-170, a known PD-L1 inhibitor, shows a binding energy of -14.49 kcal/mol, indicating a relatively strong affinity for PD-L1. However, the comparatively high standard deviation (SD = 9.38) suggests considerable variability, likely arising from the conformational flexibility of the ligand. For VISTA, the results indicate that DB00321 exhibits good affinity (-12.85 kcal/mol), with lower variability (SD = 2.76), suggesting a stable interaction. CA-170 demonstrates the strongest affinity for VISTA, with a binding energy of -17.39 kcal/mol, indicating a favorable interaction. However, its high standard deviation (SD = 8.41) suggests significant variability in interaction stability, possibly due to conformational changes in the ligand or protein. These results suggest that DB15637 and DB12867 exhibit potential inhibitory activity against PD-L1, while DB00321 shows promising inhibition against VISTA.
By gathering MM-PBSA results with the previously discussed parameters, it is evident that DB15637 and DB12867 maintain consistent compactness and stability, as reflected in their stable RMSD, RMSF, and Rg profiles.
3.9. MM-PBSA Energy Decomposition Analysis of PD-L1 and VISTA Ligand Complexes
To gain deeper insight into the molecular interactions governing ligand binding, we performed a residue-level MM-PBSA energy decomposition analysis for each PD-L1 and VISTA ligand complex (Figs. 5 and 6). This approach enabled us to identify key residues that contribute either favorably or unfavorably to the overall binding free energy.
| - | DB15637 | DB12867 | DB06744 | CA-170 |
|---|---|---|---|---|
|
MM-PBSA PD-L1 (Kcal/mol) |
-19.40 ± 4.44 | -12.85 ± 5.07 | -10.78 ± 3.82 | -14.49 ± 9.38 |
| - | DB00321 | CA-170 | ||
| MM-PBSA VISTA (Kcal/mol) | -13.57 ± 2.76 | -17.39 ± 8.41 | ||
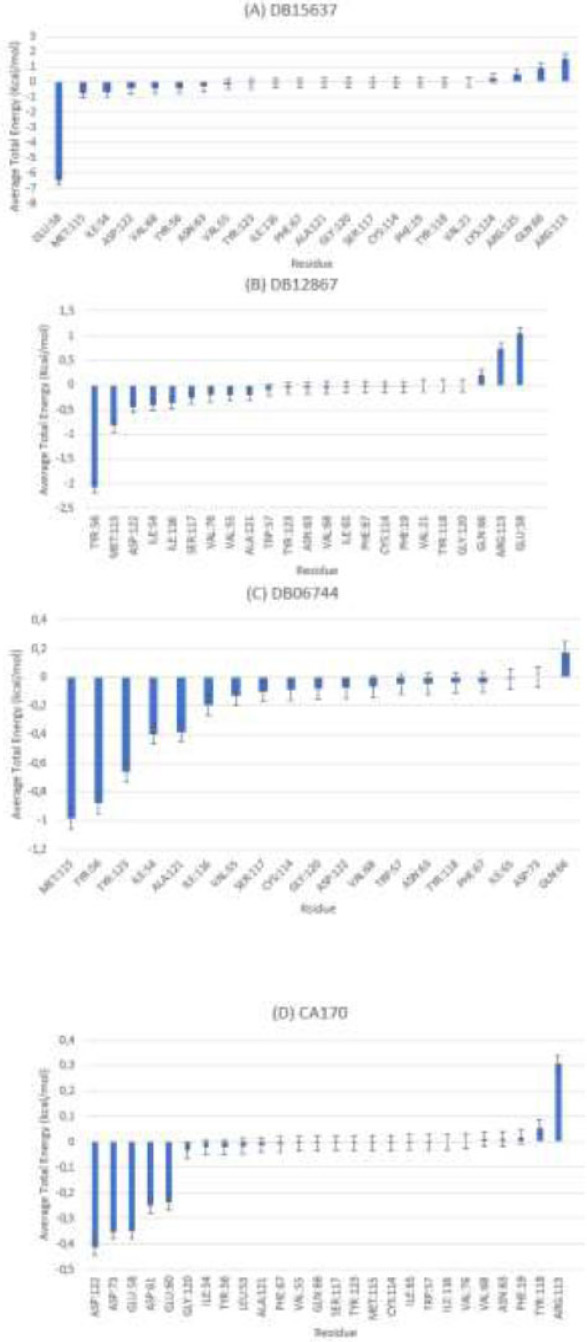
MMPBSA- PD-L1 average total energy decomposition residues (A) ligand DB15637, (B) ligand DB12867, (C) DB06744, and (D) ligand CA-170.
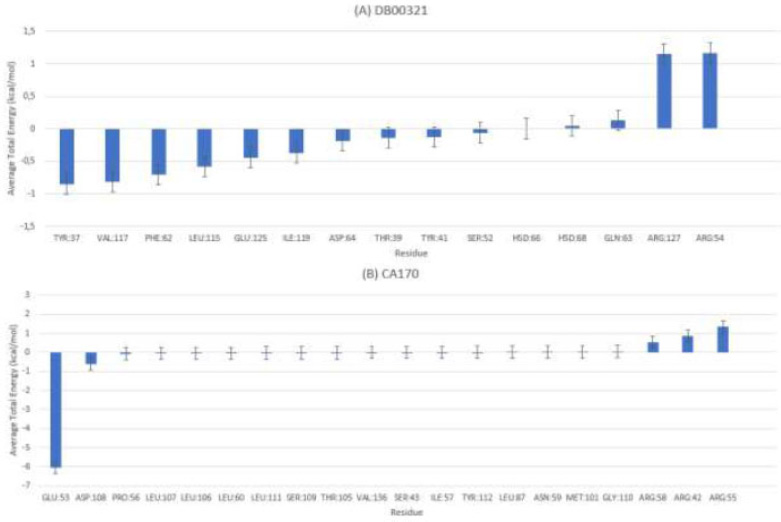
MMPBSA-VISTA average total energy decomposition residues for (A) ligand DB00321 and (B) ligand CA-170.
For the PD-L1–ligand complexes (Fig. 5), the residue-based MM-PBSA analysis revealed distinct stabilizing interactions specific to each compound. For DB15637 (Fig. 5A), the strongest stabilizing interactions involved GLU58, MET115, ILE54, and ASP122, with GLU58 contributing up to –6.5 kcal/mol. For DB12867 (Fig. 5B), key stabilizing residues included TRP56, MET115, and ASP122, with TRP56 contributing –2.2 kcal/mol. For DB06744 (Fig. 5C), favorable interactions were generally weaker; MET115, TRP56, and TRP123 contributed modestly (–0.6 kcal/mol each). For CA-170 (Fig. 5D), stabilizing contributions were limited and relatively weak, mainly involving ASP122, GLU58, and ASP73 (–0.3 to –0.4 kcal/mol each on average). These results are consistent with transient unbinding/rebinding events, particularly during the first 20–25 ns of the trajectory. However, additional analysis (Fig S2) revealed that after ~25 ns the ligand reorients and achieves a more stable conformation, as reflected by narrower binding free energy fluctuations (–5 to –30 kcal/mol).
For the VISTA-ligand complexes (Fig. 6), residue decomposition similarly highlighted key residues involved in ligand stabilization. For DB00321 (Fig. 6A), the most favorable contributions came from TYR37, VAL117, PHE62, and LEU115, each with average energy contributions ranging from –0.5 to –0.8 kcal/mol. These residues likely form critical stabilizing contacts within the VISTA binding pocket. For CA-170 (Fig. 6B), GLU53 and ASP108 contributed significantly, with GLU58 being the strongest contributor at –6 kcal/mol.
The combined analysis of MM-PBSA, RMSD, RMSF, and Rg provides a comprehensive understanding of the structural dynamics of these compounds. Although CA-170 has reached clinical trials, no conclusive data are available regarding its exact binding site. The published studies present conflicting results, and none clearly define the precise interaction site. Therefore, we performed molecular docking based on the active site of PD-L1. Our simulation revealed that the ligand identified a potential binding pocket, with ASP122, GLU58, and ASP73 emerging as key residues involved in the interaction [45-47]. Among the tested ligands, Fluzoparib (DB15637) is a PARP inhibitor approved in China for the treatment of recurrent or advanced ovarian cancer associated with BRCA mutations. By targeting DNA repair mechanisms, it enhances tumor cell death, making it a relevant candidate in oncology. Fluzoparib demonstrates remarkable stability across all measurements, making it the most promising candidate for further drug development for PD-L1. Additionally, considering its established role in PARP inhibition for ovarian cancer [48], exploring its potential repositioning as a PD-L1 inhibitor could be of great interest.
This study has also identified another potential candidate, Benperidol (DB12867), which is a butyrophenone derivative neuroleptic primarily used to treat psychoses, manic episodes, and psychomotor agitation [49]. Benperidol demonstrates strong potential with a well-balanced structural profile, suggesting it could also be repurposed for oncology applications. In contrast, DB06744 exhibits more irregular dynamic behavior, indicating the need for further optimization before it can be considered a viable therapeutic option.
These findings indicate that, although CA-170 exhibits some capacity to stabilize within the PD-L1 binding pocket after equilibration, its weak interaction energies and initial unbinding events highlight the need to identify alternative molecules with stronger and more consistent binding profiles. In this context, our proposed ligands displayed superior stability across both structural and energetic parameters, suggesting that they may represent more promising candidates for further development as PD-L1 inhibitors, while additional experimental validation will be required to confirm their therapeutic effectiveness. Additionally, the analysis of DB00321 (Amitriptyline) in the context of VISTA highlights its stable interaction with the protein. Amitriptyline is a tricyclic antidepressant mainly prescribed for major depressive disorder and neuropathic pain [50]. It demonstrates relatively consistent structural behavior, suggesting its potential as a VISTA inhibitor. However, the use of antidepressants and neuroleptics in oncology is still an emerging field, requiring further studies to understand their potential anticancer effects, optimal dosages, and possible off-target interactions.
While the repositioning of existing drugs represents a promising strategy, their clinical repurposing as immune checkpoint inhibitors should be approached with caution, given their original therapeutic indications and potential side effects. Rigorous experimental validation is essential to confirm their efficacy and safety as PD-L1 or VISTA inhibitors in the context of cancer treatment. Simultaneous inhibition of both checkpoints could potentiate the immune response against tumor cells, particularly in ovarian cancer, where PD-L1 and VISTA are highly expressed. Exploring this strategy may pave the way for novel combination therapies and ultimately improve clinical outcomes in oncology.
4. STUDY LIMITATIONS
This study is based entirely on in silico approaches, including virtual screening, molecular docking, and molecular dynamics simulations. While these computational methods provide valuable insights into the potential interactions and stability of ligand-target complexes, they cannot fully replicate the complexity of biological systems. The absence of in vitro or in vivo validation represents a limitation. Therefore, future experimental studies are necessary to confirm the biological relevance and therapeutic potential of the identified compounds.
CONCLUSION
This study has identified potential new molecules, namely DB15637, DB12867, and DB06744, as potential PD-L1 inhibitors, while DB00321 emerges as a promising VISTA inhibitor for ovarian cancer treatment. Through molecular docking analysis, 9,397 compounds from the DrugBank database were screened for their potential as PD-L1 and VISTA inhibitors. The most promising candidates were then further evaluated using 100 ns MD simulations and MM-PBSA binding free energy calculations, confirming their stability and ability to interfere with these immune checkpoints. However, further experimental validation is necessary to confirm their therapeutic potential.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: N.A.: Conceptualization; A.I.: Validation; M.H., Z.G.: Analysis and interpretation of results; N.E.: Draft manuscript; All authors reviewed the results and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| PD-1 | = Programmed Cell Death Protein 1 |
| PD-L1 | = Programmed Death-Ligand 1 |
| VISTA | = V-domain Immunoglobulin Suppressor of T cell Activation |
| OC | = Ovarian Cancer |
| PDB | = Protein Data Bank |
| MD | = Molecular Dynamics |
| RMSD | = Root Mean Square Deviation |
| RMSF | = Root Mean Square Fluctuation |
| Rg | = Radius of Gyration |
| PBC | = Periodic Boundary Conditions |
| MM/PBSA | = Molecular Mechanics Poisson Boltzmann Surface Area |
| PB | = Poisson Boltzmann |
| GB | = Generalized Born |
| SA | = Surface Area |
| HTVS | = High Throughput Virtual Screening |
AVAILABILITY OF DATA AND MATERIALS
The data sets used and/or analysed during this study are available from the corresponding author [A.I] upon request.
FUNDING
This work was carried out under National Funding from the Moroccan Ministry of Higher Education and Scientific Research to AI (PPR1). This work was also supported by a grant from the Institute of Cancer Research of the foundation Lalla Salma, Morocco, under No.591/AAP 2017 and also by a grant from the Biocodex Microbiota Foundation.
ACKNOWLEDGEMENTS
Declared none.

